赵爱月,苏云霞,傅德强
(福建医科大学 附属第二医院 肿瘤内科,福建 泉州 362000)
卵巢癌是女性最常见恶性肿瘤之一,其临床治疗尚无重大突破性进展,5年生存率保持在25%~35%[1]。虽然肿瘤免疫治疗在多个瘤种已显示出令人振奋的治疗前景[2-4],并部分应用于卵巢癌治疗领域,但仍然缺乏对卵巢癌的精准的疗效预测指标。
肿瘤免疫微环境(tumor immune microenvironment,TIME)的格局直接影响化疗和肿瘤免疫治疗的转归,与肿瘤患者预后密切相关[5-6]。CD8 T细胞浸润预示卵巢癌患者预后良好[7-8],而瘤内调节性T细胞(regulat T cell,Tregs)[7,9]或中性粒细胞[10]浸润提示预后不佳。细胞毒T淋巴细胞相关蛋白4/CD80分子(cytotoxic T-lymphocyte associated protein 4/CD80 molecule,CTLA-4/CD80)和程序性细胞死亡分子1/程序性死亡配体1(programmed cell death 1/programmed death ligand 1,PD-1/PD-L1)等免疫共信号通路主导免疫细胞的功能状态,在肿瘤免疫应答过程中发挥重要作用[11]。近年来有研究基于免疫相关基因的表达水平评估TIME格局和预测肿瘤患者的预后[11-13],但是具体机制尚未完全明朗。微小RNA(microRNAs,miRNAs)是内源性非编码RNA,通常由20~25个核苷酸组成。miRNAs可降解或抑制翻译调控靶基因表达[14],参与多种肿瘤疾病的发生、发展[15],对TIME具有调控作用。Wang等[16]在小鼠肺癌模型中发现miRNA424可以下调肺癌细胞PD-L1表达。另一项研究表明,let-7b可通过下调CD8 T细胞PD-1表达和肺癌细胞PD-L1表达来重塑TIME[17]。De Mattos-Arruda等[18]报道miRNA21可通过激活IL-6/STAT3/NF-κB和PI3K通路改变乳腺癌TIME格局。在卵巢癌中,miRNA92[19]、miRNA145[20]、miRNA424[21]等可通过不同机制上调或下调PDL1表达并改变TIME格局。然而,介导TIME形成的关键miRNA仍不清楚。
本研究以癌症基因图谱(the cancer genome atlas,TCGA)和高通量基因表达(gene expression omnibus,GEO)数据库为数据来源,拟通过生物信息学方法来筛选卵巢癌关键miRNAs及相关基因,探讨其对肿瘤免疫格局和预后的影响,阐明潜在的机制。
1.1 数据采集与预处理
所有卵巢癌患者数据均来源于TCGA(https://portal.gdc.cancer.gov/)和GEO(https://www.ncbi.nlm.nih.gov/geo/)。以TCGA数据集作为训练队列,以GEO数据集(GSE30161、GSE32062和GSE63885)作为验证队列。对所有数据进行归一化处理,使用“sva”R包去除GEO数据集的批间误差。排除总生存时间(overall survival,OS)小于1个月的患者。最终,TCGA数据集共纳入294例卵巢癌患者资料,GEO数据集共纳入393例卵巢癌患者资料。选择GSE129880、GSE117007和GSE119056数据集验证hsa-mir-7702与TIME的相关性。数据采集流程和分析均遵守TCGA和GEO的所有规定。
1.2 生物信息学分析
根据卵巢癌患者疗效与OS的线性相关性,通过“surv_cutpoint”函数计算获得PD-L1表达最佳阈值;
采用“survival”和“survminer”包进行生存差异分析;
利用“limma”包筛选差异表达基因(differentially expressed gene,DEGs),并以P<0.05,|logFC|(即|log2FC|)>0.5作为选择条件进行后续分析。通过ESTIMATE算法评估TIME格局,包括基质细胞和免疫细胞[22]。根据Bindea等[23]提供的数据集,采用单样本基因集富集分析(single sample gene set enrichment analysis,ssGSEA)算法评估免疫细胞浸润和免疫相关通路富集水平。基于拓扑重叠矩阵,采用权重基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)进行平均连锁聚类,分析某些疾病基因表达的相关性,根据基因的权重、连接度和相关系数筛选关键基因[24]。利用STRING 数据库(https://www.string-db.org/)[25]分析蛋白质-蛋白质相互作用网络(protein-protein interaction,PPI)。利用“clusterProfiler”软件包对关键基因进行基因本体论(gene ontology,GO)和京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)分析[26]。
1.3 关键基因的鉴定
Pearson相关性分析获得DEGs(包括miRNAs和mRNA)相关系数并以P<10-8,|logFC|≥0.5,频次>100为条件筛选重要DEGs,以Cor>0.5,P<0.05为条件筛选重要miRNAs,即hsa-mir-7702及其相关基因。根据WGCNA结果,选择蓝色模块中连接度Top100与PD-L1相关性Top100的基因交集,即权重基因。根据图1所示流程,筛选出关键基因。

图1 关键基因筛选流程图Figure 1 Flowchart of hub-genes identification
1.4 统计分析
应用R4.0.2软件进行统计分析,使用limma数据包中的Student t检验方法筛选DEG。KEGG中选择差异有统计学意义(P<0.05)的信号转导通路进行研究。生存分析采用Kaplan-Meier法,生存率的比较采用log-rank检验。连续变量采用t检验计算比较,分类变量通过χ2检验分析(n<5时行连续校正χ2检验)。P<0.05为差异有统计学意义。
2.1 临床特征
本研究所有患者数据均来自TCGA和GEO数据库。TCGA数据库共294例卵巢癌患者资料作为训练队列;
GEO数据库共393例卵巢癌患者资料作为验证队列,包括GSE30161数据集58例,GSE32062数据集260例,GSE63885数据集75例。卵巢癌患者的临床特征见表1。

表1 TCGA和GEO队列卵巢癌患者临床特征Table 1 Clinicopathological characteristics of patients w ith ovarian cancer in TCGA cohort and GEO cohort
2.2 卵巢癌患者PD-L1表达最佳阈值
基于卵巢癌患者OS和生存状态,计算获得PD-L1表达阈值(即第60百分位数)。根据PDL1表达阈值将所有患者分为高表达组和低表达组,生存分析结果表明,相比低表达组,高表达组具有显著生存优势(P=0.004,图2A);
在疗效方面两组未见明显差异。卵巢癌患者疗效与OS存在明显线性相关(OR=0.68,图2B)。因此,基于OS和疗效计算并获得PD-L1表达最佳阈值为1.316,即第90百分位数。根据最佳阈值分组,高、低表达组在生存(P=0.018,图2C)和疗效(P=0.006)方面均具有显著差异。验证队列分析结果显示,与低表达组比较,高表达组表现出显著生存获益(P=0.019,图2D)。

图2 卵巢癌肿瘤组织PD-L1表达最佳阈值与生存的关系Figure 2 The relationship between the optimal cutoff point of PD-L1 expression and the survival in ovarian cancer patients
2.3 PD-L1表达最佳阈值对卵巢癌TIME的评价作用
ssGSEA分析结果显示,与低表达组相比较,高表达组免疫细胞浸润更为显著,包括CD8 T细胞(CD8 positive T cells,CD8+T cells)、肿瘤浸润淋巴细胞(tumor-infiltrating lymphocytes,TILs)、T辅助细胞(T helper cells)、树突状细胞(dendritic cells,DCs)、活化树突状细胞(activated dendritic cells,aDCs)、未成熟 树突状细胞(immature dendritic cells,iDCs)、中性粒细胞(neutrophils)、巨噬细胞(macrophages)、自然杀伤细胞(natural killer cells,NK cells)、浆细胞样树突状细胞(plasmacytoid dendritic cells,pDCs)和调节性T细胞(regulatory T cells,Treg)(图3A);
高表达组免疫细胞功能更强,包括免疫检查点(check point)、T细胞共抑制(T cell co-inhibition)、T细胞共刺激(T cell co-simulation)、抗原呈递细胞共刺激(antigen-presenting cell co-simulation,APC cosimulation)、细胞溶解活性(cytolytic activity)、主要组织相容性复合体I类分子(major histocompatibility complex class I,MHC class I)、CC轴趋化因子受体(CC chemokine receptor,CCR)、I型干扰素反应(type I IFN reponse)、炎症刺激(inflammation promoting)、副炎症(parainflammation)和抗原呈递细胞共抑制(antigen-presenting cell co-inhibition,APC co-inhibition)(图3B)。ESTIMATE分析结果表明(图3C),与PD-L1低表达组比较,PD-L1高表达组具有较高免疫细胞评分(P<0.001)和ESTIMATE评分(P<0.001)。验证队列基于ssGSEA和ESTIMATE分析获得类似结论(图3 D-F)。

图3 PD-L1表达对卵巢癌肿瘤免疫格局的影响Figure 3 The effect of PD-L1 expression on tumor immune profile in ovarian cancer
2.4 DEGs的筛选结果
差异表达基因分析结果表明,共840个基因筛选为DEGs(|logFC|>0.5,P<0.05),其中,549个DEGs显著上调,291个基因显著下调(图4A)。基于DEGs的Pearson相关性分析,筛选出101个重要DEGs(P<10-8,|log FC|≥0.5,频次>100,图4B)。

图4 差异表达基因分析筛选Figure 4 The identification of DEGs
2.5 DEGs的WGCNA结果
840个DEGs的WGCNA结果(cut height=650,β=4,R2=0.9,图5 A-C)表明,有646个DEGs的蓝色模块与PD-L1表达高度相关(Cor=0.78,P=3.2×10-133,图5D-E)。
2.6 hsa-m ir-7702选择和关键基因筛选
按照图1所示流程,根据DEGs相关性分析和WGCNA结果,筛选重要miRNA和关键基因。101个重要DEGs的Cytoscape分析表明,hsa-mir-7702和75个相关DEGs具有重要作用(Cor>0.5,P<0.05,图6A)。WGCNA结果显示,蓝色模块中基因连接度和PD-L1相关性Top100基因交集后筛选出48个权重基因。7个PD-L1表达相关的miRNA分析表明,hsa-mir-7702具有较高基因连接度(connectivity=15.37),与PD-L1表达具有显著相关性(Cor=0.48,-lg P=17.56,图6B)。因此,选择 hsa-mir-7702作为关键miRNA,同时筛选出202个hsa-mir-7702相关基因(Weight≥0.3)。

图6 hsa-mir-7702和关键基因筛选Figure 6 Identification of hsa-mir-7702 and hub genes
进一步分析表明,46个基因共存于重要DEGs和权重基因,即DEG-WGCNA基因;
30个基因共存于hsa-mir-7702相关DEGs和hsa-mir-7702相关基因,即hsa-mir-7702互作基因。DEGWGCNA基因和hsa-mir-7702互作基因交集分析获得15个关键基因,包 括 TIGIT(T cell immunoreceptor with Ig and ITIM domains,携有Ig和ITIM 结构域的T细胞免疫受体)、IL2RG(interleukin 2 receptor subunitgamma,白介素2受体γ亚基)、ICOS(inducible T cell costimulator,诱导T细胞共刺激因子)、SH2D1A(SH2 domain containing 1A,携有1A 的SH2域)、SLAMF6(SLAM family member 6,SLAM 家族成员6)、GZMA(granzyme A,颗粒酶A)、P2RY10(P2Y receptor family member 10,P2Y受体家族成员10)、SLAMF8(SLAM familymember 8,SLAM家族成员8)、CD247(CD247 molecule,CD247分子)、PDCD1LG2(programmed cell death 1 ligand 2,程序性细胞死亡1配体2)、CYTIP(cytohesin 1 interacting protein,细胞蛋白1相互作用蛋白)、RHOH(ras homolog family member H,ras同源家族成员H)、LCK(LCK proto-oncogene,LCK原癌基因)、STX11(syntaxin 11,突触融合蛋白11)和CXCR6(C-X-Cmotif chemokine receptor 6,CX-C轴趋化因子受体6)(图6C)。
2.7 hsa-m ir-7702在卵巢癌TIME的作用
根据hsa-mir-7702表达中位值将卵巢癌患者分为高表达组和低表达组,无监督聚类分析结果表明,与低表达组相比较,高表达组展现出更显著免疫细胞浸润和更强的免疫应答能力(图7A)。验证队列包括GSE119056、GSE115019和GSE129880数据集中,也有类似现象(图7B)。
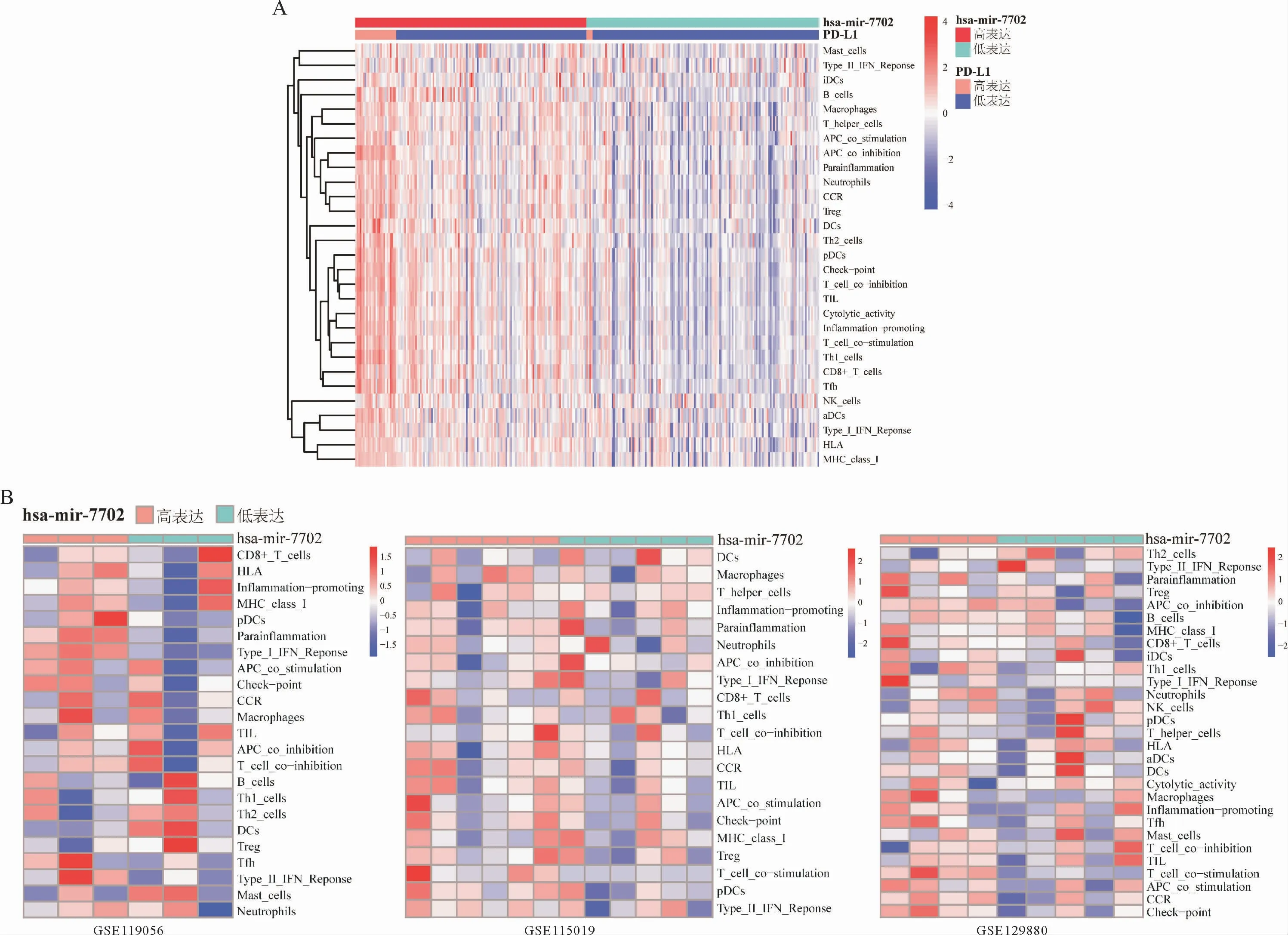
图7 hsa-mir-7702表达对卵巢癌肿瘤免疫格局的影响Figure 7 Tumor immune profile in high-and low-expression groups according to median of hsa-mir-7702 in TCGA cohort and validation cohort
2.8 关键基因功能富集和PPI分析
KEGG分析表明,关键基因主要涉及免疫相关信号通路,包括PD-L1表达和PD-1检查点通路、自然杀伤细胞介导细胞毒作用、T细胞受体信号通路(图8A)。GO分析表明,关键基因主要介导免疫相关生物学功能(图8A)。其中,主要生物学过程(biological processes,BP)包括细胞-细胞黏附、T细胞活化正调控和T细胞共刺激;
主要细胞成分(cellular components,CC)包括质膜外侧面结构和免疫突触;
主要分子功能(molecular function,MF)包括C-C趋化因子结合、C-C趋化因子受体活化、T细胞受体结合、免疫受体活化等。PPI分析结果显示,15个关键基因与PD-L1存在直接或间接的相互作用(confidence=0.6,图8B)。
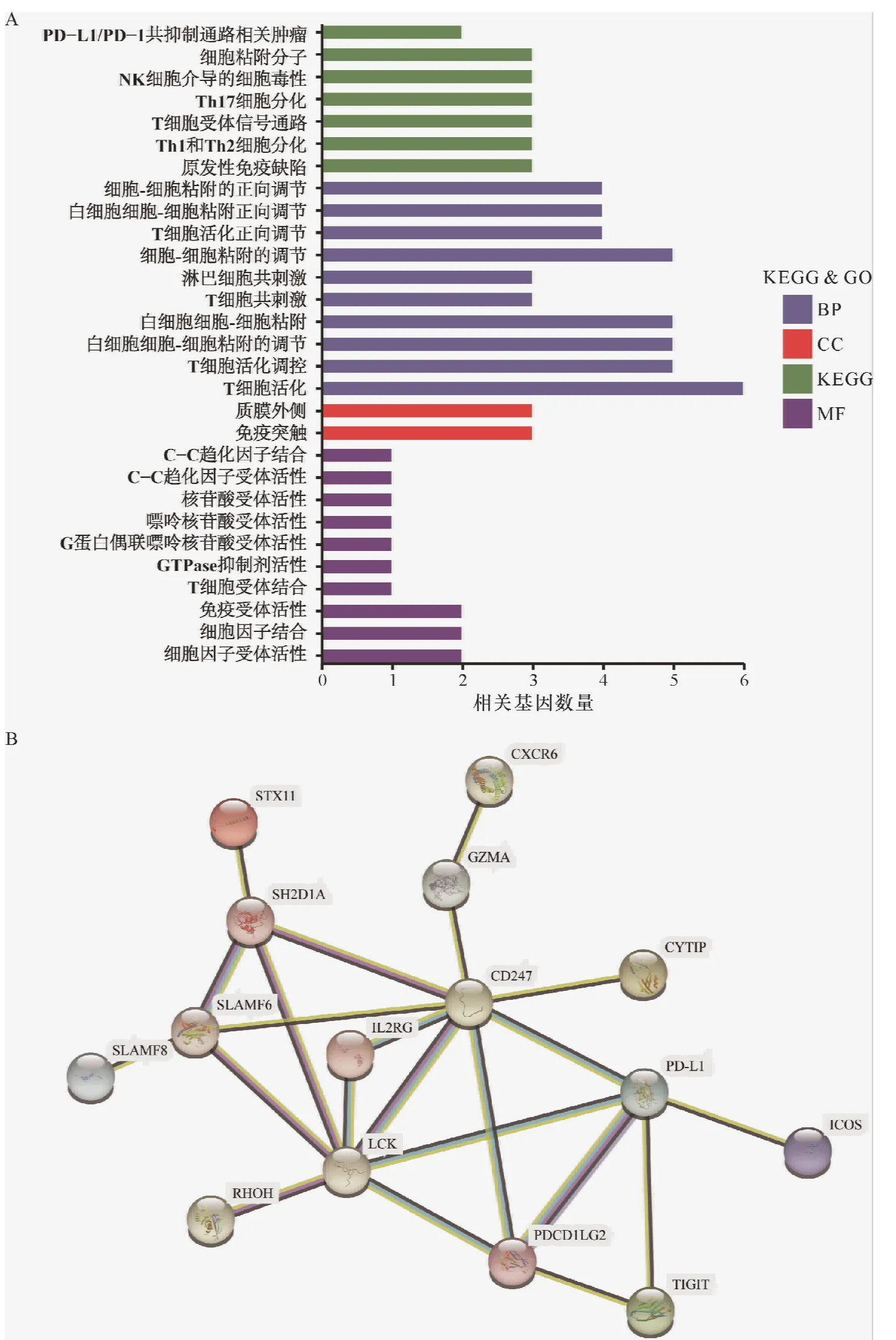
图8 关键基因的功能富集分析和PPI分析Figure 8 Function analysis and PPIanalysis of15 hub genes
2.9 关键基因的生存分析
根据关键基因表达中位值分为高、低表达组,生存分析结果表明,训练队列6个关键基因(TIGIT、IL2RG、ICOS、SH2D1A、SLAMF6和GZMA)表达与患者生存显著相关(图9A);
验证队列13个关键基因(TIGIT、SH2D1A、LCK、PD-L1、GZMA、CXCR6、ICOS、IL2RG、SLAMF8、STX11、CD247、RHOH和PDCD1LG2)表达与患者生存密切相关(图9B)。

图9 15个关键基因与卵巢癌预后的关系Figure 9 Survival curves of 15 hub genes grouped by themedian of each gene expression
TIME是肿瘤细胞所处周围环境的免疫格局,包含淋巴细胞、固有免疫细胞、中性粒细胞、成纤维细胞和内皮细胞。大量研究证实,TIME与多种肿瘤疾病预后密切相关[27],包括卵巢癌[7,28]、非小细胞肺癌[29]、三阴性乳腺癌[30]和结直肠癌[31]。本研究中,ssGSEA分析和ESTIMATE分析的结果证实了PD-L1表达对卵巢癌TIME的影响,高表达组具有更强的免疫应答能力,并提示患者良好的预后。
本研究旨在探讨miRNAs对卵巢癌TIME的影响,而辨别不同的肿瘤免疫格局非常重要。PD-L1是最重要的免疫相关基因之一,与T细胞免疫功能密切相关,大量研究将其作为判断肿瘤免疫状态的评价指标。然而,截至目前,PD-L1表达最佳阈值尚不清楚。本研究基于卵巢癌患者疗效和OS获得PD-L1表达最佳阈值,即第90百分位数。生存分析结果高度支持PD-L1表达最佳阈值的适用性和可靠性。
DEG筛选和WGCNA分析是甄别关键基因的重要方法。本研究结合两种分析方法,根据基因相关性、连接度、频次和权重筛选重要miRNA和关键基因。DEGs相关性分析和WGCNA结果均高度支持hsa-mir-7702在卵巢癌患者肿瘤免疫格局形成过程的重要性。15个hsa-mir-7702相关的关键基因主要包括ICOS、PDCD1LG2、TIGIT、SH2D1A、IL2RG、CXCR6、STX11、SLAMF6、P2RY10、RHOH、CD247、GZMA、SLAMF8、CYTIP和LCK。已经明 确,ICOS、PDCD1LG2、TIGIT 和SLAMF6是重要的免疫相关共信号分子[32]。PPI分析提示,15个关键基因存在直接或间接的相互作用。进一步的KEGG分析和GO分析结果提示15个关键基因主要涉及肿瘤免疫相关信号通路,并介导T细胞免疫应答的所有环节,包括致敏、激活、增殖、迁移、识别和杀伤。因此,推测hsamir-7702可能通过调控15个关键基因介导卵巢癌TIME形成。
许多研究证实了miRNA对卵巢癌TIME的调控作用,如miRNA424可通过抑制PD-L1和CD80表达,恢复T细胞免疫应答能力,扭转化疗药物耐药[21];
miRNA145可在体外通过结合c-Myc转录因子下调卵巢癌细胞PD-L1的表达[20];
miRNA92可通过抑制LATS2表达介导卵巢癌细胞PD-L1表达上调[19]。目前,尚未有hsa-mir-7702对卵巢癌TIME影响的相关报道。一项涉及结直肠癌的研究表明,hsa-mir-7702可通过靶向作用于转录衔接因子1(transcriptional adaptor 1,TADA1)而抑制肿瘤细胞迁移和侵袭[33];
另一项关于恶性黑色素瘤研究发现,hsa-mir-7702表达与PD-L1呈正相关[34]。此外,hsa-mir-7702在多个生物信息学相关的肿瘤风险预测模型均有报道,包括结直肠癌[35]、黑色素瘤[36]、膀胱癌[37-38]和乳腺癌[39]。本研究ssGSEA分析结果提示,hsamir-7702表达上调有利于促进卵巢癌患者的肿瘤免疫应答能力,包括免疫功能和免疫细胞浸润。尽管验证队列样本量有限,但是也表现出同样趋势。此外,PD-L1和hsa-mir-7702均高表达的患者具有更强的免疫应答能力。基于以上结果,本研究认为hsa-mir-7702可能通过调控15个关键基因而对卵巢癌肿瘤免疫格局产生影响。
关键基因通过各种机制参与肿瘤疾病发生、发展并影响患者预后。本研究发现训练队列6个关键基因与卵巢癌患者生存密切相关,包括IL2RG、TIGIT、ICOS、SH2D1A、SLAMF6和GZMA;
验证队列13个关键基因与患者预后显著相关。表明15个关键基因对卵巢癌预后转归具有重要作用,但由于恶性肿瘤具有异质性,在不同群体中所主导作用可能存在一定差异。
本研究支持hsa-mir-7702对卵巢癌肿瘤微环境的免疫调节作用,但仍存在一定局限性,首先,本课题基于生物信息学方法进行数据分析,方法较为单一;
其次,部分验证队列样本量小,所得结论可能存在一定误差。因此,后续研究将针对hsa-mir-7702深入挖掘。
作者贡献声明
赵爱月:数据收集和整理,文献检索,撰写论文;
苏云霞:数据收集和整理;
傅德强:提出研究思路、统计分析数据,论文审阅和修改。
利益冲突声明
本研究未受到企业、公司等第三方资助,不存在潜在利益冲突。
猜你喜欢队列卵巢癌关键硝酸甘油,用对是关键中老年保健(2022年1期)2022-08-17新形势下深化改革开放的关键一招江苏钢铁(2022年9期)2022-07-02miR-181a在卵巢癌细胞中对顺铂的耐药作用昆明医科大学学报(2022年1期)2022-02-28高考考好是关键中学生数理化(高中版.高考理化)(2021年6期)2021-07-28队列里的小秘密小学生学习指导(低年级)(2020年4期)2020-06-02基于多队列切换的SDN拥塞控制*软件(2020年3期)2020-04-20在队列里军营文化天地(2018年2期)2018-12-15丰田加速驶入自动驾驶队列产品可靠性报告(2017年7期)2017-09-05CXXC指蛋白5在上皮性卵巢癌中的表达及其临床意义中国癌症杂志(2015年4期)2015-12-09卵巢癌脾转移的临床研究进展肿瘤预防与治疗(2015年2期)2015-09-26